Syntax
spectra = powspec(obj,QA)
spectra = powspec(___,Name,Value)
Description
spectra = powspec(obj,QA) calculates powder averaged spin wave spectrum
by averaging over spheres with different radius around origin in
reciprocal space. This way the spin wave spectrum of polycrystalline
samples can be calculated. This method is not efficient for low
dimensional (2D, 1D) magnetic lattices. To speed up the calculation with
mex files use the swpref.setpref('usemex',true) option.
spectra = powspec(___,Value,Name) specifies additional parameters for
the calculation. For example the function can calculate powder average of
arbitrary spectral function, if it is specified using the specfun
option.
Example
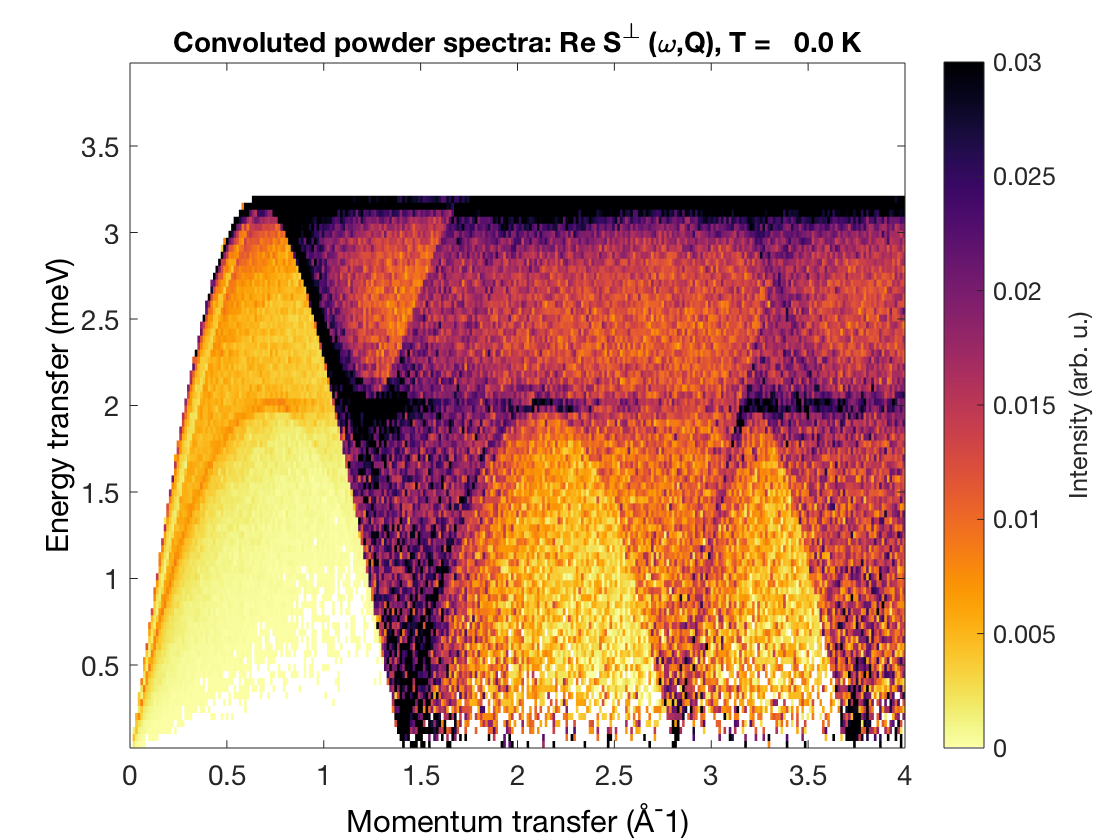
Using only a few lines of code one can calculate the powder spectrum of the triangular lattice antiferromagnet (\(S=1\), \(J=1\)) between \(Q=0\) and 3 Å\(^{-1}\) (the lattice parameter is 3 Å).
tri = sw_model('triAF',1);
E = linspace(0,4,100);
Q = linspace(0,4,300);
triSpec = tri.powspec(Q,'Evect',E,'nRand',1e3);
sw_plotspec(triSpec);
Input arguments
obj- spinw object.
QA- Vector containing the \(Q\) values in units of the inverse of the length unit (see spinw.unit) with default unit being Å\(^{-1}\). The value are stored in a row vector with \(n_Q\) elements.
Name-Value Pair Arguments
specfun- Function handle of a solver. Default value is
@spinwave. It is currently tested with two functions:spinw.spinwavePowder average spin wave spectrum.spinw.scgaPowder averaged diffuse scattering spectrum.
nRand- Number of random orientations per
QAvalue, default value is 100. Evect- Row vector, defines the center/edge of the energy bins of the
calculated output, number of elements is \(n_E\). The energy units are
defined by the
spinw.unit.kBproperty. Default value is an edge binlinspace(0,1.1,101). binType- String, determines the type of bin, possible options:
'cbin'Center bin, the center of each energy bin is given.'ebin'Edge bin, the edges of each bin is given.
Default value is
'ebin'. 'T'- Temperature to calculate the Bose factor in units
depending on the Boltzmann constant. Default value taken from
obj.single_ion.Tvalue. 'title'- Gives a title to the output of the simulation.
'extrap'- If true, arbitrary additional parameters are passed over to the spectrum calculation function.
'fibo'- If true, instead of random sampling of the unit sphere the Fibonacci
numerical integration is implemented as described in
J. Phys. A: Math. Gen. 37 (2004)
11591.
The number of points on the sphere is given by the largest
Fibonacci number below
nRand. Default value is false. 'imagChk'- Checks that the imaginary part of the spin wave dispersion is smaller than the energy bin size. Default value is true.
'component'- See sw_egrid for the description of this parameter.
The function also accepts all parameters of spinw.spinwave with the most important parameters are:
'formfact'- If true, the magnetic form factor is included in the spin-spin
correlation function calculation. The form factor coefficients are
stored in
obj.unit_cell.ff(1,:,atomIndex). Default value isfalse. 'formfactfun'- Function that calculates the magnetic form factor for given \(Q\) value.
value. Default value is
@sw_mff, that uses a tabulated coefficients for the form factor calculation. For anisotropic form factors a user defined function can be written that has the following header:F = formfactfun(atomLabel,Q)where the parameters are:
Frow vector containing the form factor for every input \(Q\) valueatomLabelstring, label of the selected magnetic atomQmatrix with dimensions of \([3\times n_Q]\), where each column contains a \(Q\) vector in \(Å^{-1}\) units.
'gtensor'- If true, the g-tensor will be included in the spin-spin correlation function. Including anisotropic g-tensor or different g-tensor for different ions is only possible here. Including a simple isotropic g-tensor is possible afterwards using the sw_instrument function.
'hermit'- Method for matrix diagonalization with the following logical values:
trueusing Colpa’s method (for details see J.H.P. Colpa, Physica 93A (1978) 327), the dynamical matrix is converted into another Hermitian matrix, that will give the real eigenvalues.falseusing the standard method (for details see R.M. White, PR 139 (1965) A450) the non-Hermitian \(\mathcal{g}\times \mathcal{H}\) matrix will be diagonalised, which is computationally less efficient. Default value istrue.
'fid'- Defines whether to provide text output. The default value is determined
by the
fidpreference stored in swpref. The possible values are:0No text output is generated.1Text output in the MATLAB Command Window.fidFile ID provided by thefopencommand, the output is written into the opened file stream.
'tid'- Determines if the elapsed and required time for the calculation is
displayed. The default value is determined by the
tidpreference stored in swpref. The following values are allowed (for more details see sw_timeit):0No timing is executed.1Display the timing in the Command Window.2Show the timing in a separat pup-up window.
The function accepts some parameters of [spinw.scga] with the most important parameters are:
'nInt'- Number of \(Q\) points where the Brillouin zone is sampled for the integration.
Output Arguments
spectra- structure with the following fields:
swConvThe spectra convoluted with the dispersion. The center of the energy bins are stored inspectra.Evect. Dimensions are \([n_E\times n_Q]\).hklASame \(Q\) values as the inputhklA.EvectContains the bins (edge values of the bins) of the energy transfer values, dimensions are \([1\times n_E+1]\).paramContains all the input parameters.objThe clone of the inputobjobject, see spinw.copy.